

REGENERATE.
REVITALIZE.
RECOVER.
Are you on a journey to find a healthier, stronger and more vibrant version of you? Welcome to the future of your health. With our high-tech treatments and team of experts to guide you, together we can unlock your purest and most powerful form.
Get started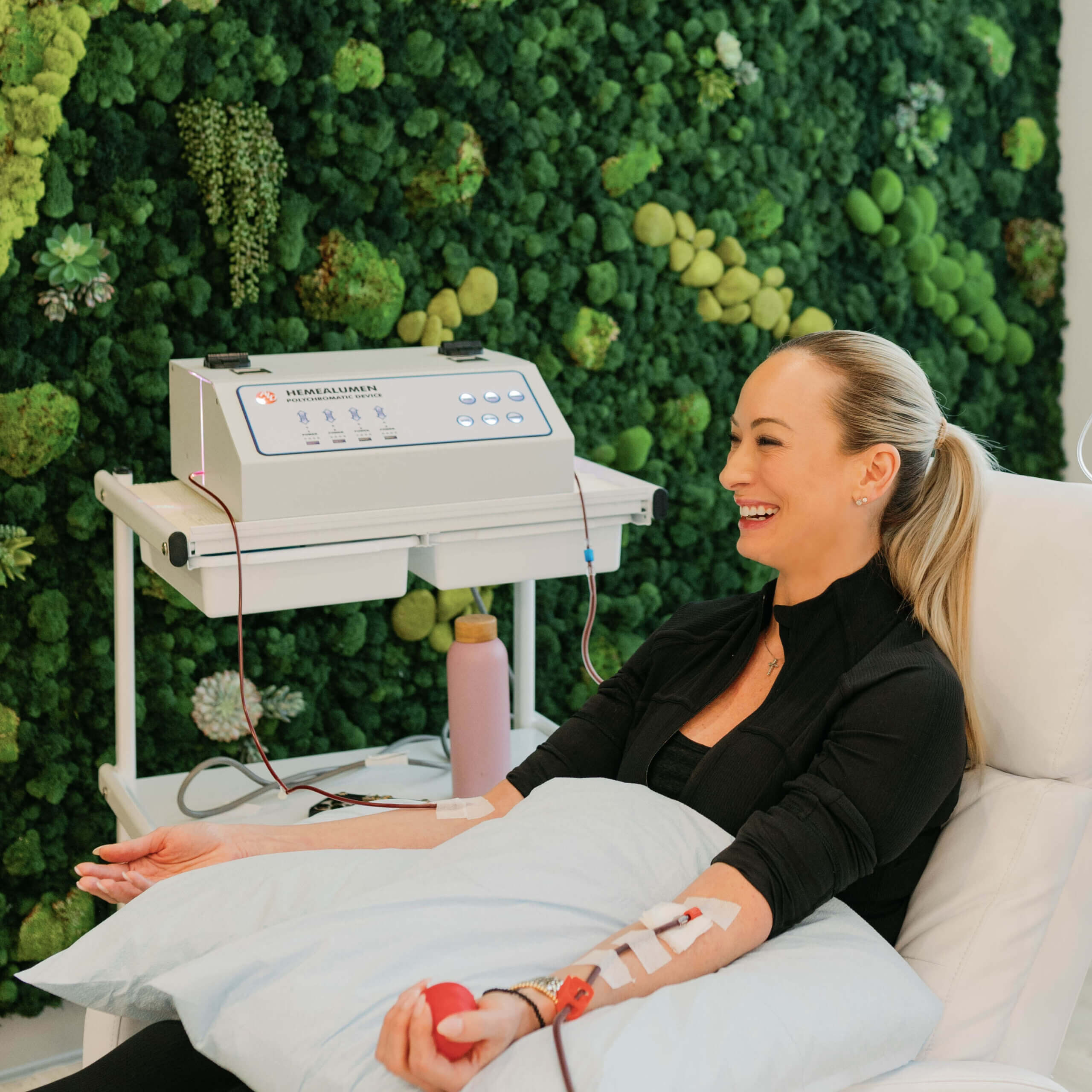














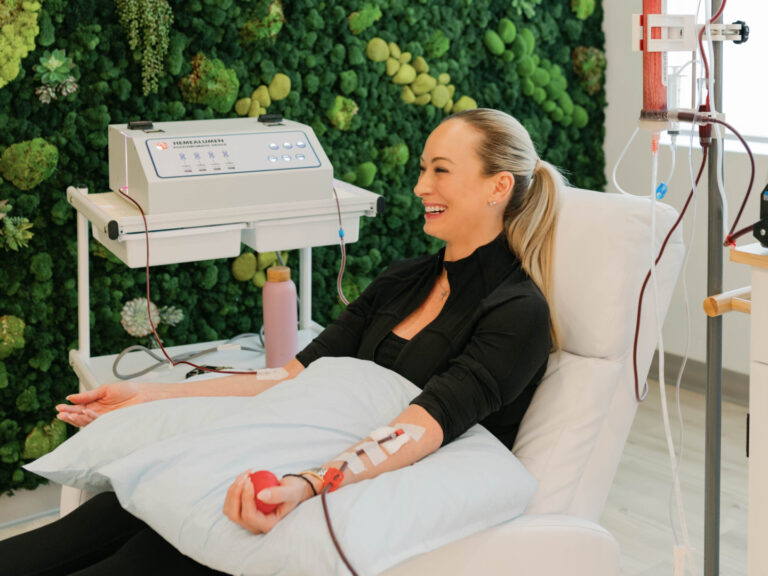


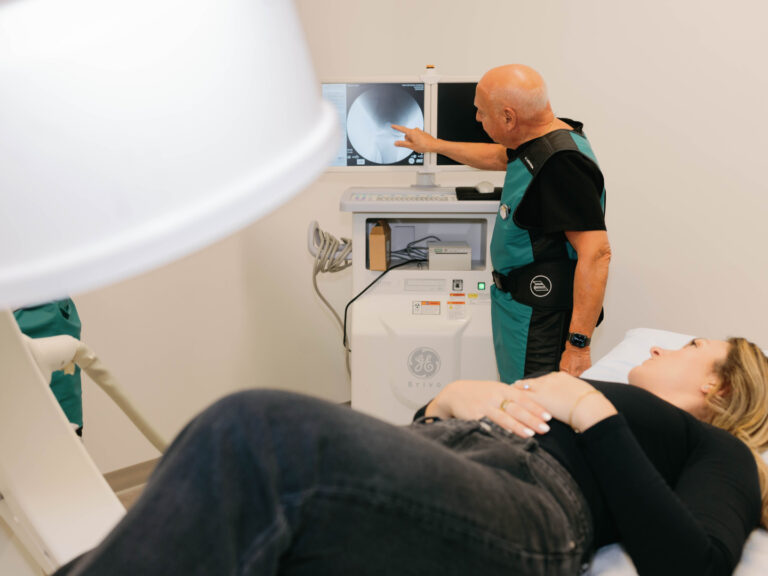
Dr. Purita is a pioneer of Orthopedic Regenerative Cell and Platelet Rich Plasma (PRP) treatments. Educated at the prestigious Georgetown University, University of Florida Medical Center and the University of Miami-Jackson Memorial Hospital, Dr. Purita is board certified in Orthopedic Surgery, Pain Management and Regenerative Medicine.